 +1-2404726069 (U.S.)
+1-2404726069 (U.S.) 
 0
0In the early 1980s, researchers discovered that the sensitivity of chromatin to DNase I was related to gene transcription. When a gene is transcriptionally active, the chromatin regions containing the gene are significantly more sensitive to DNase I degradation. More than 100-fold higher than transcriptionally inactive regions, these locations are known as DNase I hypersensitive sites. So what is the specific principle of DNase I hypersensitive sites? How it be identified?
1. What are DNase I hypersensitive sites?
2. What is the process for DNase I hypersensitivity sites?
3. Techniques to identify DNase I hypersensitive sites
4. What are the transcriptional regulation features of DNase I hypersensitive sites?
1. What are DNase I hypersensitive sites?
DNase I hypersensitive sites, as the name suggests, are sites that are sensitive to DNase I cleavage. Specifically, DNase I hypersensitive sites are regions of chromatin spanning hundreds of base pairs that are less methylated and highly sensitive to DNase I. In specific regions of the genome, chromatin loses its condensed structure. The chromatin regions where genes are being transcribed or have transcription potential are loosely structured. DNA is thus exposed, increasing its likelihood of being degraded by non-specific nucleases, such as DNase I. DNase I hypersensitive sites can enrich transcription factors and important enzymes, and have transcriptional regulatory functions.
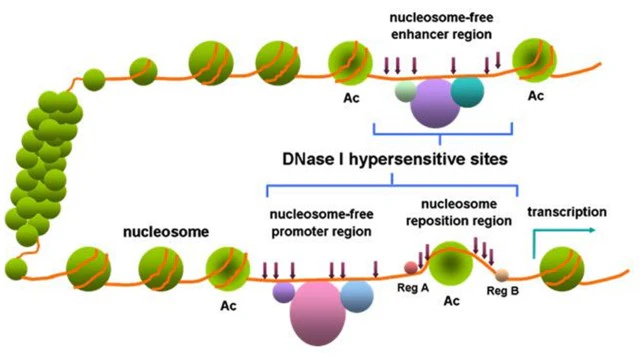
Figure 1. DNase I hypersensitive sites within chromatin[1]
DNase I hypersensitive sites can be used as markers for regulatory DNA regions. These regions have been shown to reflect multiple types of cis-regulatory elements, including promoters, enhancers, insulators, silencers, and site control regions. And high-throughput measurements of these regions were obtained by DNase I digestion coupled with high-throughput sequencing (DNase-seq).
With the development of high-throughput technology, especially large-scale sequencing technology, there are currently more studies on yeast, Drosophila, and human genomes. First, DNase I is used to degrade fragments to obtain fragments, and then chip hybridization or large-scale sequencing is used to analyze the results. A systematic bioinformatics analysis was performed to obtain the identification results of DNase I hypersensitive sites at the genome-wide level.
To systematically search for DNase I hypersensitive sites, the project of the Encyclopedia of DNA Elements (ENCODE) utilizes a new generation of high-throughput technologies. For the first time, a large-scale map of human DNase I hypersensitive sites has been mapped. A total of 125 DNase I hypersensitive sites in the human cell and tissue types are depicted in the whole genome, which contributes to a comprehensive understanding of the regulatory mechanism of the human genome. By analyzing the resulting data, ENCODE's project identified 2,890,742 high-confidence DNase I hypersensitivity sites, each activated in one or more cell types. Among them, 970,100 DNase I hypersensitivity sites was owned by a single cell type, 1,920,542 DNase I hypersensitivity sites was activated in two or more cell types, and 3,692 DNase I hypersensitivity sites was detectable in all cells.
2. What is the process for DNase I hypersensitivity sites?
To identify DNase I hypersensitivity sites, the first step is to extract the nucleus, digest it with DNase I, and separate large fragments by pulsed-field gel electrophoresis. A linker containing a type II restriction endonuclease Mme I site was then ligated by T4 DNA ligase and end-labeled with biotin. The ligated product was digested with Mme I, another adapter was ligated at the nick, and then PCR was performed to amplify the ligated product. The PCR products were recovered and a library was constructed. The Illumina high-throughput sequencer was used for sequencing. Finally, the sequencing results were analyzed by bioinformatics to identify DNase I hypersensitivity sites.
In the bioinformatics analysis process of DNase I hypersensitivity sites, the first step is to remove the linker sequence during library construction and then compare the linker-removed data with the target genome. The comparison results were analyzed using Formaldchyde Assisted IsoJation of Regulatory Elements high-throughput sequencing (F.Seq) software. Then output the wig format file to facilitate the use of subsequent genome visualization tools. If the result of the region identified by F.Seq satisfies the condition of FDR<0.01, the site is a DNase I hypersensitivity site. DNase I hypersensitivity sites were correlated with expression profile data. Finally, the obtained results were displayed using the genome visualization tool GBrowser. The established DNase I hypersensitivity sites can be integrated with genomic information in public databases, which facilitates subsequent in-depth comparative analysis.
3. Techniques to identify DNase I hypersensitive sites
Benefiting from the improvement of high-throughput sequencing technology, some new technologies have been applied to detect DNase I hypersensitivity sites. Examples include DNase-seq and chromatin immunoprecipitation with high-throughput sequencing (CHIP-seq).
CHIP is a relatively mature technology for analyzing protein binding sites. Its main principle is to first use physical or chemical methods to destroy the chromatin of cells and expose DNA samples containing transcription factor proteins. The samples are filtered, precipitated, cross-linked, reverse-cross-linked, aligned to DNA fragments, and then subjected to high-throughput sequencing of these DNA fragments. After the sequencing result is obtained, the algorithm is compared with the base sequence of the human genome to obtain the position information of the gene in the genome. Finally, the information on the protein binding site is obtained.
Although CHIP-seq technology is relatively mature, it can accurately identify the binding sites of regulatory proteins. But the disadvantage is that if the sequenced fragment is repeated many times in the genome, it is impossible to infer whether it is a binding site. There are disadvantages such as high sequencing cost, difficult matching of specific enzymes, and long time-consuming. In addition, the specific enzymes of some transcription factors are difficult to find, which hinders the CHIP-seq technology to complete the sequencing of all transcription factor sites in the whole genome. DNase-seq can effectively avoid these problems. It has low data cost and no specific enzymes. It can measure a large range of gene regions at one time, and the detection accuracy can reach a single base, while CHIP-seq technology may be due to peak findings. The problem with the algorithm is that there is a deviation of several bp.
DNase-seq technology combines DNA footprinting and high-throughput sequencing technology. After the protein is combined with DNA, it can protect the binding site from being degraded by DNase I, and the protein can be determined by determining the DNA fragments left after DNase I degradation. The region that binds to DNA, this method is called DNA footprinting. To read more about DNA footprinting click on the link. This method can precisely identify the binding regions of transcription factors, outperforming traditional CHIP methods. The main principle of action of DNase-seq is to use DNase I to hydrolyze DNA strands bound to transcription factor proteins. The specific three-dimensional structure of the DNA strand will protect the DNA to a certain extent, and the specific degree of effect will be affected by the protein structure. Therefore, different transcription factors will form different shear shapes due to different shapes, thereby forming a set of characteristic patterns. The size of each position value is related to the sequencing depth of each experiment, but the overall shape shows a pattern of differences due to the protein structure of the transcription factor. Since the experimental principle used by DNase-seq technology is the cleavage properties of DNase I enzymes, there is no specific transcription factor specificity. If the experiment is performed on the whole gene, all protein binding sites in the whole genome can be detected at one time, which makes up for the inadequacy of CHIP-seq technology.
The main technical process of DNase-seq technology is as follows: the first step is to add the reaction system to the sample to be tested and add a certain amount of DNase I enzyme to accurately grasp the reaction time. It is beneficial to the extraction of information, but if the reaction time is too long, the fragment will be too small, and then the accuracy of the alignment to the genome cannot be guaranteed. In the second step, special biotin primers are added to the treated DNA fragments, PCR amplification is performed on them, and then the ssDNA library is constructed. In the third step, the genes in the library are sequenced by high-throughput sequencing, and the obtained gene sequences are compared with the reference genome to obtain the position of DNase I digestion.
According to the ENCODE Consortium, DNase-seq sequencing is the core method for the recent identification of human epigenomes. It does not rely on antibodies or epitope tags and can analyze the genomic distribution of a large number of proteins in a single experiment. It is suitable for a range of cell types. However, mapping nucleosome localization using DNase-seq sequencing is somewhat complicated because DNase I cuts nucleosomal DNA with a resolution of 10 bp. At the same time, DNase-seq technology also has some problems to be solved. First, the results of DNase-seq are based on the differences in footprints generated by the three-dimensional structural differences of different proteins. These differences are directly reflected in the DNase-seq data itself, so it is necessary to design a reasonable algorithm to distinguish and improve the recognition accuracy. This is also an important technical difficulty for DNase-seq technology to identify binding sites. Second, because the DNase I enzyme has a base bias, this bias is an unwanted factor in the patterning of the three-dimensional structures of protein. Therefore, it is necessary to find a reasonable method to remove the influence of this factor.
4. What are the transcriptional regulation features of DNase I hypersensitive sites?
Transcription factors act by binding to transcriptional regulatory elements on chromatin, which are usually located in or around chromatin DNase I hypersensitive sites. Using CHIP-seq technology, the binding sites to DNA in certain groups of transcription factors were determined. When comparing to the maps of DNase I hypersensitive sites, the results showed a high correlation, indicating that the coordinated binding of certain factors is related to chromatin remodeling.
DNA methylation patterns silence transcription, thereby inhibiting the transcriptional expression of genes. -C-phospho-G-(CpG) methylation is closely associated with transcriptional silencing, and this methylation causes chromatin rearrangements, which condense and inactivate during transcription. Data are suggesting that methylation patterns parallel cell-selective chromatin accessibility as a result of passive deposition of transcription factors following their separation from regulatory DNA. Methylated CpGs that fall within DNase I hypersensitive sites hinder the association of transcription factors and DNA, inhibiting chromatin accessibility. When the degree of methylation increases, the degree of chromatin openness decreases.
Nucleosomes can block the binding of transcription factors to cis-acting elements. The lack of nucleosomes means that the chromatin structure is in an open or loose state, and transcription factors can enter and then bind to cis-acting elements to initiate gene transcription. The trimethylation of histone third subunit lysine 4 (H3K4me3) is associated with transcriptional activity, and this modification occurs in the adjacent nucleosomes at the transcription start site and can relax chromatin structure. Such histone modifications can be used as promoter markers to map elements in the human genome. DNase I hypersensitive sites, which are transcription factor binding sites, act by binding transcription factors to enhance the openness of chromatin.
Some transcriptional regulatory elements near or distal to RNA polymerase binding sites on chromatin can cooperate with transcriptional regulators to determine the initiation and efficiency of gene transcription. The ENCODE project identified 578,905 distal DNase I hypersensitive sites that were highly associated with at least one target promoter. A model combining distal DNase I hypersensitive sites and specific promoters in different cell types was confirmed. And the model map of the distal DNase I hypersensitive sites promoter was drawn. It was confirmed that the distal DNase I hypersensitive sites bind to specific promoters to make the distal DNase I hypersensitive sites function as enhancers, thereby enhancing gene expression.
The above is the relevant content about DNase I hypersensitive sites. To read more about DNase I, please click this link.
References
[1] Wang YM, Zhou P, Wang LY, Li ZH, Zhang YN, Zhang YX. Correlation between DNase I hypersensitive site distribution and gene expression in HeLa S3 cells. PLoS One. 2012;7(8):e42414. (IF 3.752)
