 +1-2404726069 (U.S.)
+1-2404726069 (U.S.) 
 0
0Next-generation sequencing (NGS), or second-generation sequencing (of course, there are also third-generation sequencing and fourth-generation sequencing) are familiar words to biology researchers even if some of you have already processed NGS experiments. But, do you know what exactly NGS, or, what exactly NGS studies? Here, this essay will describe these answers in detail.
We will focus on Second-Generation Sequencing-related technologies because they are the biggest family among all the NGS technologies. Second-generation sequencing-related technologies can be divided into three main categories: DNA research-related technologies, RNA research-related technologies, and epigenetic research-related technologies.
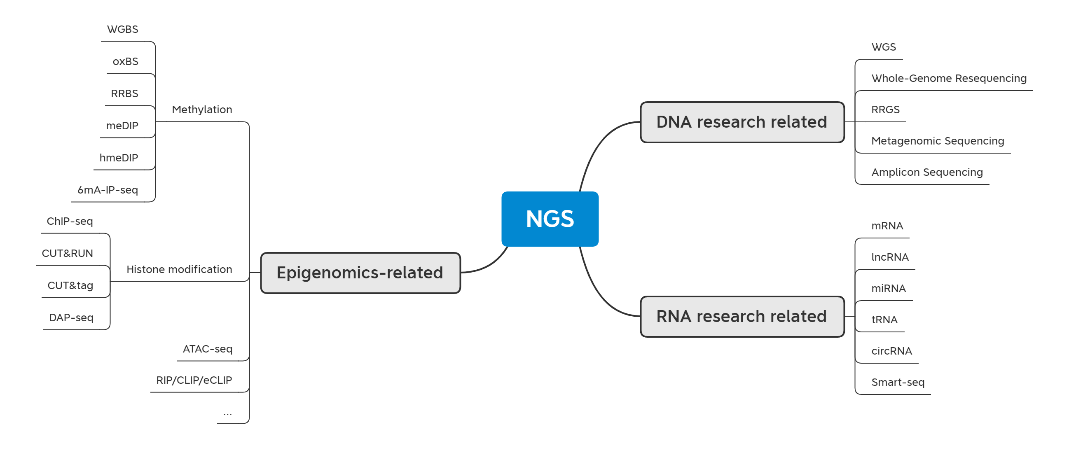
Fig1. NGS atlas
1. DNA research-related technology
2. RNA research-related technology
3. Epigenomics-related techniques
4. Other Modules
1. DNA research-related technology
The main steps of DNA research technology: DNA extraction, DNA fragmentation, DNA library construction, sequencing, and data analysis.
It seems that the steps of DNA-related research technology are not that difficult, however, if you think DNA research is very simple, then you are too young and too simple.
First of all, among all the DNA research0related technologies, Whole Genome Sequencing (WGS) is the one that has to be mentioned. From the completion of the human genome project, human beings began to enter the genome era, and genomic information of different species was reported one after another. The bioinformatics analysis methods of the genome are also daily changes. The story of the genome is renewed every day. So, we will not go into the details here. But we should know that all the research on second-generation sequencing. Before entering this field, we still need to confirm whether the species to be studied has a genome. Of course, someone may say that analysis without reference genome is now available. It is true, but the analysis without a reference genome is always the last choice, and when there is a reference genome, please use the way with a reference genome to analyze the data.
Based on genome sequencing, there are also Whole-Genome Resequencing and Reduced-Representation Genome Sequencing (RRGS). These techniques are currently mainly applied to the construction of genetic atlases, germplasm resource identification, plant and animal genetic breeding, structural variation detection, trait gene localization, population evolution analysis, etc. They also derive some proprietary names for analysis such as BSA, SNP, Indel, SV, QTL, GWAS, etc. In addition to these, there is a Metagenome (or, microbial environmental genome) which is used to research environmental samples such as soil, feces, gut, and water, or pathogenic samples. Metagenome is also developed based on single sample genomic research techniques. There are also population-based technologies that study the sequence of one or some parts of genes such as DNA barcoding, macro-barcoding, etc. or Amplicon Sequencing such as 16S/18S/ITS sequencing, functional genome sequencing, eclipse analysis, etc.
|
Categories |
Product Name |
Cat# |
Specification |
|
Mechanical library construction |
12197ES24/96 |
24/96 T |
|
|
Enzyme-based library construction |
12205ES24/96 |
24/96 T |
|
|
Mechanical library construction |
Hieff NGS™ Ultima Pro DNA Library Prep Kit for MGI® V2 (Inquire) |
13312ES16/96 |
16/96 T |
|
Enzyme-based library construction |
Hieff NGS™ OnePot Pro DNA Library Prep Kit for MGI® (Inquire) |
13322ES16/96 |
16/96 T |
2. RNA research-related technology
The main steps of RNA-related research techniques are the same, which are RNA extraction, mRNA capture (rRNA removal), RNA library construction, sequencing, and data analysis.
The main objects of RNA research are mRNA, miRNA, circRNA, lncRNA, and tRNA, and the library construction methods are different according to the characteristics of the objects. For example, mRNA has a poly-A tail, so oligo dT beads are used to capture poly-A; while miRNA, because of its small fragments, is usually detected with non-denaturing gel running gel and then cut gel to recover. The circRNA library building is divided into two cases, one is to build a library of circRNA only, in this case, the linearized RNA needs to be removed first, and then to build a circRNA library. The other way to build lncRNA library is by removing rRNA, which can get mRNA, lncRNA, and also some circRNA. To study tRNA, we need to specifically capture the hairpin structure of tRNA, then add the demethylation step, and build tRNA library.
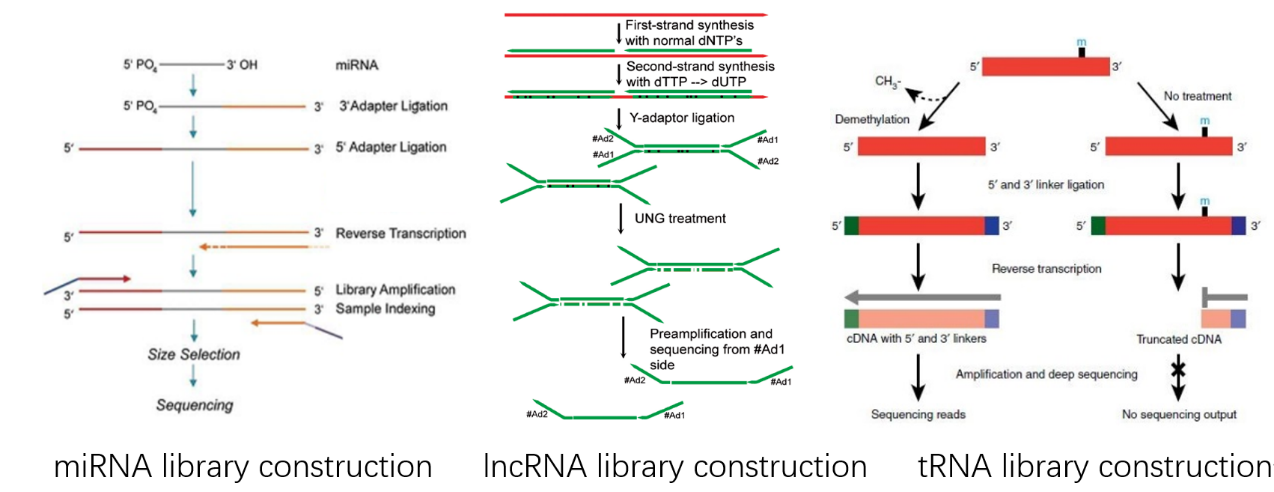
Fig2: Non-coding RNA library construction workflow
Here, we would like to talk about a special transcriptome study, which is single-cell RNA-seq. Single cells are usually prone to failure using conventional library building methods because of the small amount of RNA. To achieve single-cell RNA library construction, we need to make the whole experiment in one tube as much as possible, instead of the conventional need to keep changing EP tubes during the whole experimental process. Currently, magnetic beads are mostly used to achieve one-tube operation. This is because when building a library, the mRNA library is usually constructed.
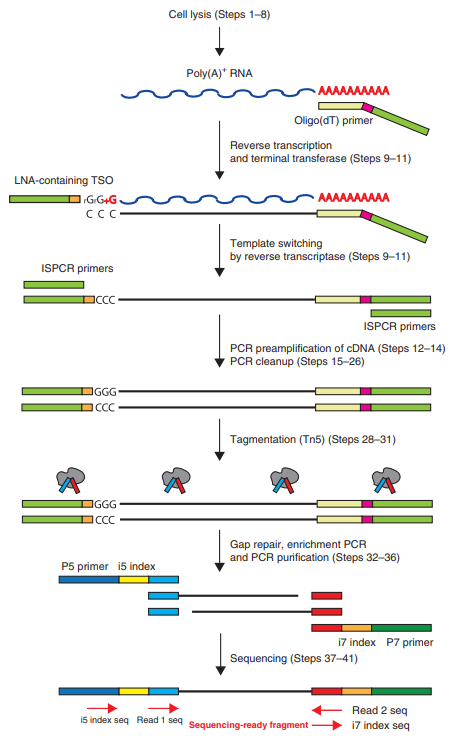
Fig3: Smart-seq2 workflow [1]
|
Categories |
Product name |
Cat# |
Specification |
|
mRNA libraries and RNA strand-specific libraries |
Hieff NGS™ Ultima Dual-mode mRNA Library Prep Kit (for Illumina and MGI) |
12309ES24/96 |
24/96 T |
|
LncRNA+mRNA conventional libraries and strand-specific libraries |
Hieff NGS™ Ultima Dual-mode RNA Library Prep Kit (for Illumina and MGI) (Inquire) |
12308ES24/96 |
24/96 T |
3. Epigenomics-related techniques
Epigenomics focuses on the mechanisms by which nucleic acids do not change, but phenotypes do. At present, epigenomics research is divided into four major parts: methylation research, histone modification research, chromatin remodeling research, and non-coding RNA regulation research. And there are very many types of techniques involved.
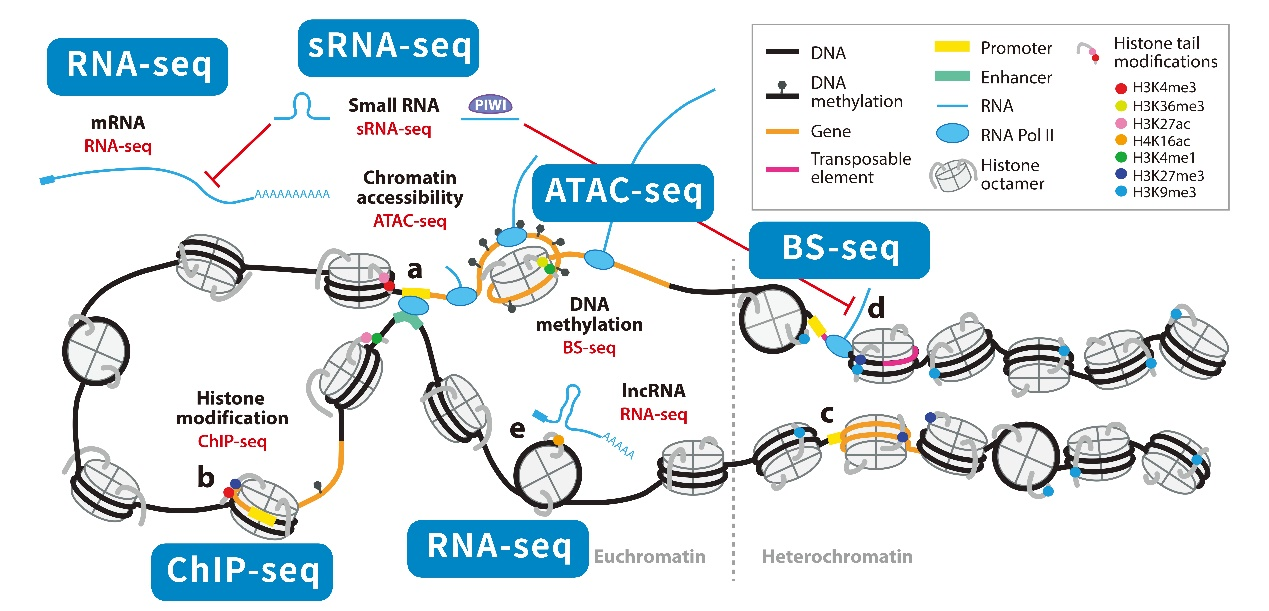
Fig4. Epigenomic atlas
3.1 Methylation studies
Methylation studies are divided into two major categories, one is DNA methylation and the other is RNA methylation. DNA methylation is currently studied with two major modification types, 5mC and 6mA, while the types of techniques used for 5mC studies are WGBS, RRBS, oxBS, meDIP-seq and hmeDIP-seq, and for 6mA studies are 6mA-IP-seq. RNA methylation studies also work on 5mc and 6mA, and the techniques used are meDIP and meRIP, too.
Different methylation study techniques use different technical principles. WGBS`s principle is by using sulfite treatment in which unmethylated C bases are converted to U and later to T in PCR amplification. this method is the most classical genome-wide methylation study method and the gold standard for methylation studies, which can obtain genome-wide methylation modifications. The principle of RRBS is using methylation-sensitive enzymes to cleave genomic DNA, and the capture regions are mainly CpG islands. oxBS technique is used to study hydroxymethylation modification. During the experiment, two libraries need to be constructed, one is the conventional WGBS, and the other library first converts intermediate states of methylation modification states by enzymes. To be specific, 5hmC is oxidized to 5fc by enzymatic oxidation, after which all these intermediate states of methylation can be converted to U when treated with sulfite, and then converted by the bioinformatic method. Another major category is the use of specific antibodies for targeted capture, such as meDIP, hmeDIP, and 6mA-IP-seq and meRIP.
3.2 Histone modification studies
The study of histone modifications focuses on chromatin regulation and gene expression regulation in which various different histone modifications are involved. Histone modification techniques are those of DNA-protein interactions represented by ChIP-seq. These types of techniques are ChIP-seq, CUT&RUN, and CUT&tag.
3.3 Chromatin remodeling studies
Chromatin remodeling studies focus on chromatin regulation and regulation of gene expression by chromatin remodeling factors. The types of techniques studied are consistent with histone modifications, also ChIP-seq, CUT&RUN, and CUT&tag.
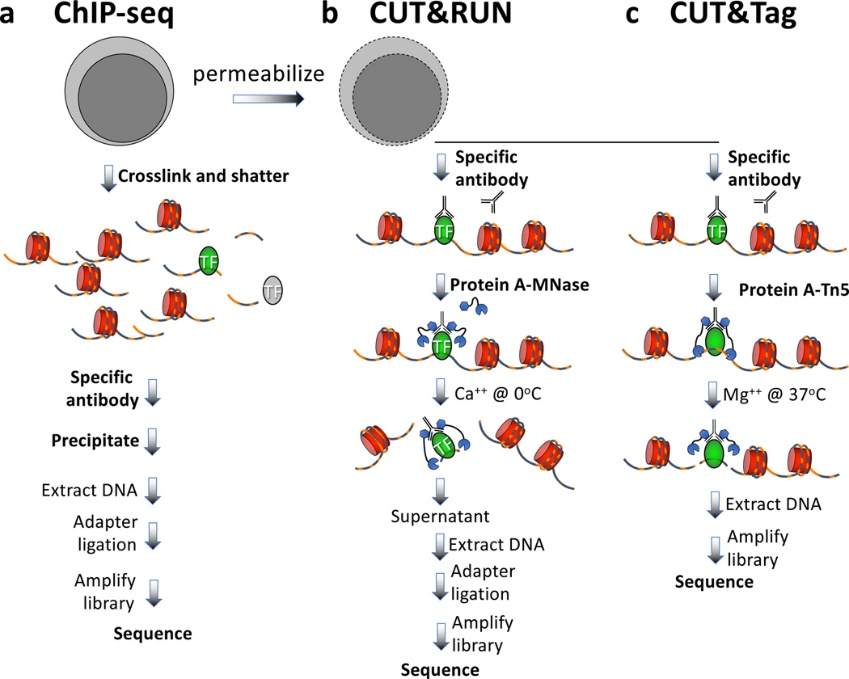
Fig5. DNA-protein interaction techniques workflow
In addition to these DNA-protein interaction techniques described above, there is also DAP-seq, which also studies DNA-protein interactions, but it is an in vitro experiment and is suitable for DNA-protein interaction studies of some non-model organisms. The advantage of this experiment is that no antibody is needed and no transgenic system is required, but because it is an in vitro experiment, it is easy to produce many false positives.
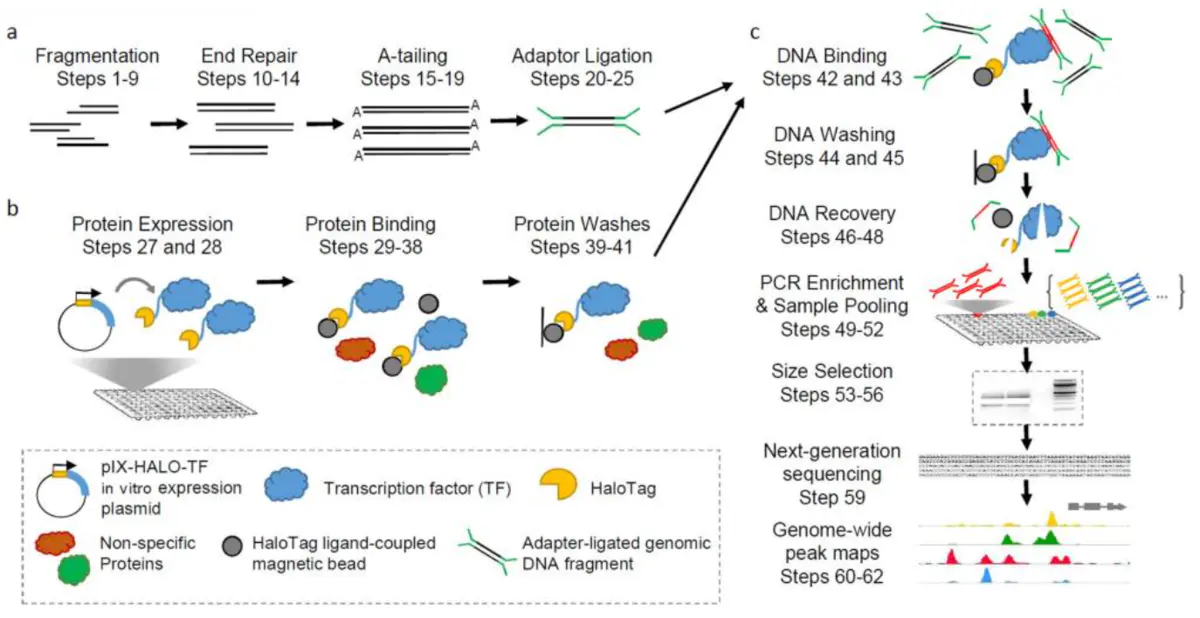
Fig6. DAP-seq workflow [2]
There is also the chromatin open research technique, ATAC-seq, which focuses on the use of Tn5 transposase to cleave chromatin open regions and later analyze these open regions' DNA after library building.
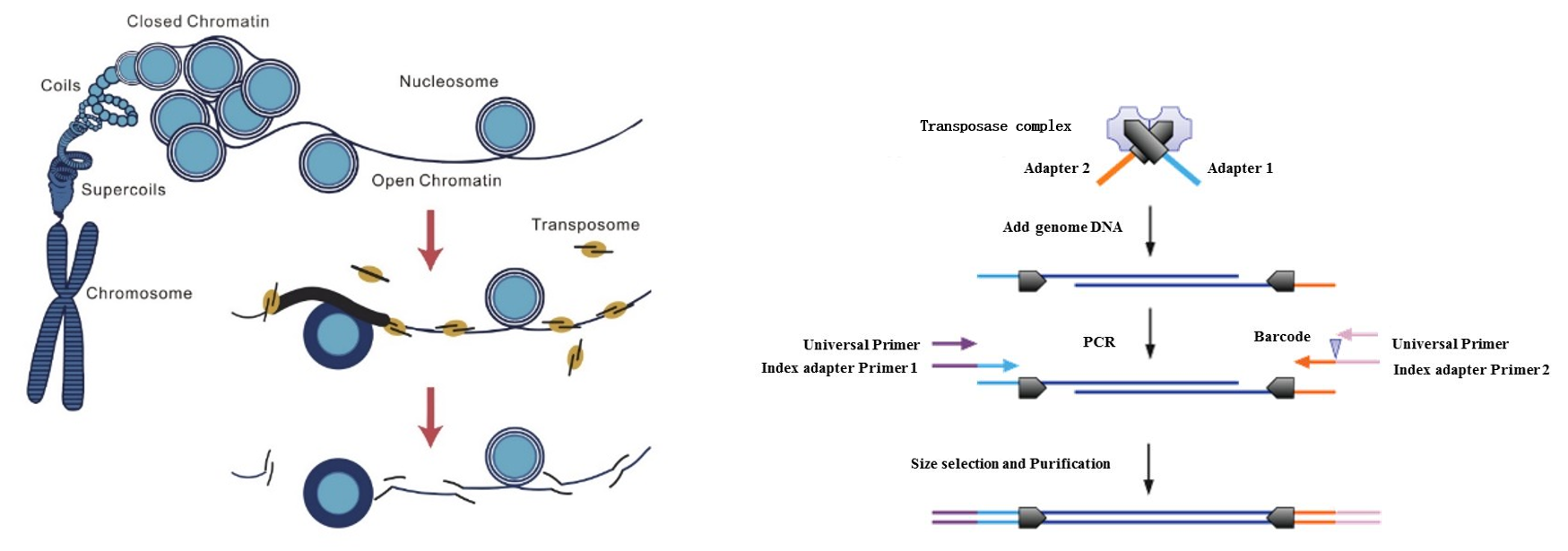
Fig7. ATAC-seq library construction workflow [3]
3.4 Non-coding RNA regulation
For non-coding RNA regulation, besides the conventional RNA library construction, there are also RNA-protein interactions technology studies. The most classical study is about RNA-binding proteins (RBPs). RNA pull-down and RIP-seq/CLIP/miCLIP/eCLIP are the main techniques to achieve the research purpose.
RNA pull-down is used to pull down unknown interacting proteins with known RNA, and then pulled-down proteins are identified by mass spectrometry. Usually, we can choose RNA pull-down and RIP/CLIP/miCLIP/eCLIP simultaneously to perform forward and reverse validation of the RNA and RBP to be verified.
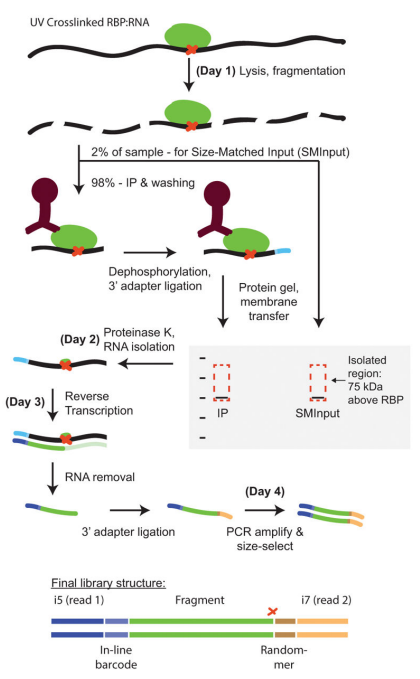
Fig8. eCLIP workflow [4]
4. Other Modules
|
Categories |
Product Name |
Cat# |
Specification |
|
Short adaptor & pair-end index |
Hieff NGS™ 384 Dual Index Primer Kit for Illumina, Set 1/Set 2 (Inquire) |
12412ES02 |
96×2 T |
|
Long adaptor & single-end index |
Hieff NGS™ Complete Adapter Kit for Illumina, Set 1/ Set 2 (Inquire) |
13519ES04/16 13520ES04/16 |
48×4 T/16 T |
|
UDI adaptor & pair-end index |
12404ES01 12405ES01 |
12×2 T |
|
|
single-end barcode & long adaptor -MGI |
Hieff NGS™ Complete Adapter Kit for MGI (Inquire) |
13360ES02/04/96 13361ES02/04/96 13362ES02/04/96 |
8×2/4 /100 T |
|
Short adaptor & UMI UDB primer-MGI |
Hieff NGS™ Dual UMI UDB Adapter Kit for MGI (Inquire) |
13367ES02/04 13368ES02/04 |
48×2 T/4 T |
|
Short adaptor & UDB primer-MGI |
Hieff NGS™ Unique Dual Index Primer Kit for MGI (Inquire) |
13451ES02/04 13452ES02/04 13453ES02/04 13454ES02/04 |
48×2 T/4 T |
|
DNA purification |
12601ES |
/ |
|
|
DNA ligase |
Hieff™ Novel T4 DNA Ligase (400 U/µL) (Inquire) |
10298ES40/42 |
40/400 KU |
|
Long adaptor & single-end index |
Hieff NGS™ Complete Adapter Kit for Illumina, Set 1/ Set 2 (Inquire) |
13519ES04/16 13520ES04/16 |
48×4 /16 T |
|
CDI adaptor & pair-end index |
Hieff NGS™ RNA 384 CDI Primer for Illumina Set1/ Set2 (Inquire) |
12414ES02 |
96×2 T |
|
Short adaptor & CDI primer-RNA-MGI |
Hieff NGS™ RNA 384 CDI Primer for MGI Set1/ Set2 (Inquire) |
13365ES02 13366ES02 |
96×2 T |
|
Including DNA fragmentation, end repair and A-addition steps |
Hieff NGS™ OnePot Pro DNA Fragmentation Reagent (Inquire) |
12619ES24/96 |
24 T/96 T |
|
Including end repair and A-addition steps |
Hieff NGS™ Fast-Pace End Repair/dA-Tailing Module (Inquire) |
12608ES24/96 |
24 T/96 T |
|
rRNA removal (plant) |
12254ES24/96 |
24 T/96 T |
|
|
rRNA removal (Human rRNA & ITS/ETS) |
12257ES24/96 |
24 T/96 T |
|
|
Human globin depletion probe |
Globin mRNA Depletion Probe (Human) (Inquire) |
12806ES24/96 |
24/96 T |
|
Highly efficient and fidelity U-base-resistant DNA amplification enzyme |
Hieff Canace™ Uracil+ High-Fidelity DNA Polymerase (Inquire) |
10145ES60/76 |
100 /500 U |
|
Library amplification enzyme module |
Hieff Canace™ Pro Amplification Mix for NGS (Inquire) |
12624ES24/96 |
24 T/96 T |
Regarding reading:
Key enzymes involved in NGS library construction
DNase I and Their Applications in Biomedicine
Reverse Transcriptase Selection
References:
[1] Picelli S, Faridani O R, Bjrklund S K, et al. Full-length RNA-seq from single cells using Smart-seq2[J]. Nature Protocols, 2014, 9(1):171-181.
[2] Ecker, Joseph, R, et al. Mapping genome-wide transcription-factor binding sites using DAP-seq[J]. Nature Protocols Erecipes for Researchers, 2017.
[3] Buenrostro J D, Wu B, Chang H Y, et al. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide[J]. Current Protocols in Molecular Biology, 2015, 109(1).
[4] Nostrand E L V, Pratt G A, Shishkin A A, et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP)[J]. Nature Methods.
